男性,56 岁,进行性双上肢无力 6 年。
初始症状为右上臂无力,3 个月后出现左上臂无力,此后双上臂无力逐渐加重伴肌肉萎缩、肉跳,不能抬高、梳头,双手尚可持物,无肢体麻木,无言语含糊。
既往史无特殊,父母身体健康,否认有遗传病家族史。
体格检查:神清,言语清晰,对答切题,定向力、记忆力和计算力正常,无舌肌萎缩及纡颤,舌肌顶颊肌力 5,其余颅神经未见阳性体征。
颈软,双上臂和肩胛带肌明显萎缩,双上肢可见束颤(视频),双侧肩关节内收、外展肌力 2 级,肘关节屈、伸肌力 3 级,双上肢远端肌力 4 级,双下肢肌力 5 级,双上肢肌张力减低,双下肢肌张力正常。
双上肢腱反射对称+,双下肢膝反射++,四肢深浅感觉检查正常,双侧 Hoffman 征阴性,双侧掌颌反射阴性,双侧 Babinski 征、chaddock 征阴性。
定位诊断思路见下图
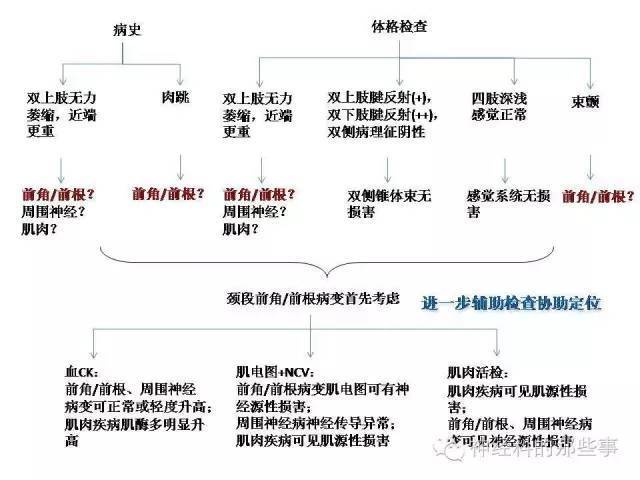
病人血CK正常;神经传导测定提示双侧正中神经、尺神经运动传导CMAP波幅降低,余上、下肢周围神经运动和感觉传导均正常;
针极肌电图示双上肢神经源性损害(双侧三角肌、肱二头肌、伸指伸肌、小指展肌可见大量自发电位及高波幅、宽时限的MUP)。
根据患者肌电图表现,考虑前角/前根可能性较大,但周围神经病变尚不能完全排除。
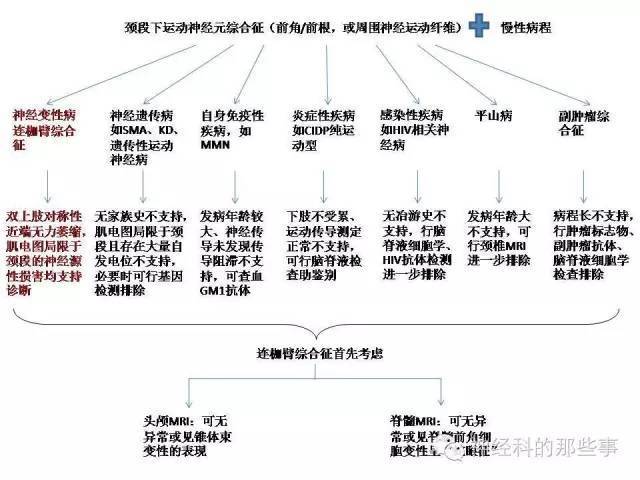
进一步行血生化、甲状腺功能、自身免疫抗体、HIV抗体、GM1抗体、肿瘤标志物、副肿瘤抗体均正常,脑脊液常规、生化、细胞学正常,双下肢SEP正常。
肺部CT、腹部彩超等检查未见明显异常,颅脑、颈椎MRI未见明显异常。
最终诊断:连枷臂综合征(flail arm syndrome, FAS)
基于本病例的问题:
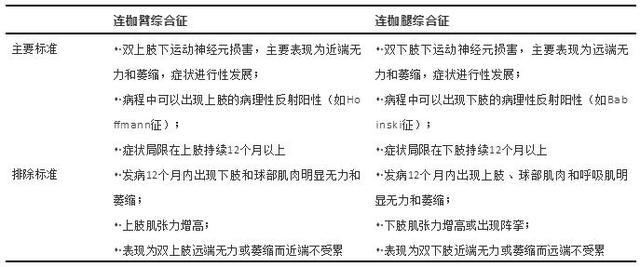
Q1: 连枷臂综合征(FAS)和连枷腿综合征(FLS)如何诊断?
FAS和FLS的诊断主要基于患者的临床表现,目前多采用2009年Wijesekera提出的诊断标准:
Q2: 连枷臂综合征和连枷腿综合征有上运动神经元损害吗?
FAS 和 FLS 主要表现为局限于上肢或者下肢的下运动神经元损害,但在病程中可以出现 Hoffmann 征、Babinski 征等病理反射。
研究发现即便是不存在锥体束征的的 FAS 和 FLS 患者,神经电生理的表现也与经典的 ALS 相似,如皮质和周围兴奋性增高,或者 MEP 的中枢运动传导时间延长;
病理上可见到典型的上运动神经元受累的病理特征,如脊髓胸段和腰段的皮质脊髓侧束有髓纤维的广泛脱失。
Q3: 连枷臂综合征和上肢起病的ALS临床上有何区别?
FAS 和上肢起病的 ALS 临床上存在一些差异:FAS 患者肌无力和萎缩主要位于上肢近端,而上肢起病的 ALS 患者肌无力以上肢远端为主;上肢起病的 ALS 患者肌束颤动明显较 FAS 患者更常见;FAS 病理征少见而 ALS 多见;FAS 患者自发病到第二个体区出现功能障碍的时间要明显比上肢起病的 ALS 长(平均分别为 34.3 个月和 12.3 个月)。
此外,神经电生理研究还发现 FAS 患者不存在 ALS 患者典型的分裂手现象(split-handphenomenon)。
分裂手现象是指患者拇短展肌和第一骨间背侧肌更早出现肌萎缩无力且受累程度更重,而小指展肌相对保留,在神经电生理上表现为拇短展肌/小指展肌 CMAP 波幅比和第一骨间肌/小指展肌 CMAP 波幅比明显降低。
然而,需要指出,少数 FAS 在病程中也可出现病理反射,而且近 2/3 FAS 患者在出现第二个体区的功能障碍前肌电图即可出现第二个、第三个体区的神经源性损害,因此,临床上 FAS 和上肢起病的 ALS 单次就诊也很难完全区分开来。
Q4: 连枷臂综合征的鉴别诊断包括哪些?
虽然 FAS 的典型表现为双上肢近端对称性无力、萎缩,但一项对 42 例 FAS 患者的研究发现仅 25% 患者起病时表现为双侧对称性无力、萎缩,而 75% 患者起病时仅表现为单侧上肢无力、萎缩。
仅 24% 患者起病时表现为单纯近端无力或萎缩,而 40% 患者以单纯远端无力,36% 患者以远端近端同时无力起病,由于在疾病的早期症状多数不典型,误诊率可以高达 55%。
连枷臂综合征的鉴别诊断包括平山病、脊髓性肌萎缩症、多灶性运动神经病、遗传性运动神经病、CIDP 纯运动型、HIV 相关神经病、面肩肱型肌营养不良、颈椎病、双侧皮质分水岭梗死,和脊髓梗死等。
Q5: 连枷臂综合征和连枷腿综合征的预后如何?
总体而言,连枷臂综合征和连枷腿综合征的预后要明显好于经典型 ALS。
国外一项对 135 例 FAS 和 75 例 FLS 的研究发现 FLS 和 FAS 的中数生存期(分别为 69 个月和 61 个月)明显比上肢起病和延髓起病的 ALS 患者(分别是 34 个月和 27 个月)长,这四种临床表型的 5 年生存率分别为 63.9%、52%、20% 和 9.3%。
国内樊东升教授对 126 例 FAS 的研究发现 FAS 的中数生存期(97 个月)明显比肢体起病和延髓起病的 ALS 患者(分别是 72 个月和 48 个月)长,三种临床表型的 5 年生存率分别为 58%、49% 和 37%。
参考文献
1.Wijesekera LC, Mathers S, Talman P,Galtrey C, Parkinson MH, Ganesalingam J, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology.2009;72:1087-94.
2.Chen L, Zhang B, Chen R, Tang L, Liu R, Yang Y, et al. Natural history and clinical features of sporadic amyotrophic lateral sclerosis in China. J NeurolNeurosurg Psychiatry. 2015;86:1075-81.
3.Hubers A, Hildebrandt V, Petri S,Kollewe K, Hermann A, Storch A, et al. Clinical features and differential diagnosis of flail arm syndrome. J Neurol. 2016;263:390-5.
4.Jawdat O, Statland JM, Barohn RJ, Katz JS, Dimachkie MM. Amyotrophic Lateral Sclerosis Regional Variants (Brachial Amyotrophic Diplegia, Leg Amyotrophic Diplegia, and Isolated Bulbar Amyotrophic Lateral Sclerosis). Neurologic clinics. 2015;33:775-85.
5.Yoon BN, Choi SH, Rha JH, Kang SY, Lee KW, Sung JJ. Comparison between Flail Arm Syndrome and Upper Limb Onset Amyotrophic Lateral Sclerosis: Clinical Features and Electromyographic Findings. Experimental neurobiology. 2014;23:253-7.
6.Yang H, Liu M, Li X, Cui B, Fang J, Cui L. Neurophysiological Differences between Flail Arm Syndrome and Amyotrophic Lateral Sclerosis. PLoS One. 2015;10:e0127601.
