接诊了一例 MRI 提示双侧纹状体萎缩的患者,让我们跟着病例一起回顾下双侧纹状体受累的疾病。
姿势步态异常 3 年的儿童
患者女,8 岁,因「姿势、步态异常 3 年,发作性肢体抽搐 1 个月」入院。
5 岁时逐渐出现行走不稳,行走脚尖着地,身体易向后跌倒(惊吓时易出现)。左肘关节屈曲,行走时左上肢连带动作消失,左手指变形,呈「兰花指样」。颈部向左偏及前倾。
1 个月来反复发作性四肢抽搐伴意识障碍,每次发作持续约 1-2 分钟恢复正常。既往运动发育较同年龄儿童落后。否认药物、毒物接触史。否认家族遗传性疾病史。

患者双足内翻畸形,左肘关节屈曲,左手「兰花指样」畸形。
查体:
体型瘦小。神志清楚,言语匮乏(仅简单言语交流),智能减退,颅神经查体阴性。
四肢肌力 V 级。四肢肌张力增高,双下肢为甚。双侧膝反射、跟腱反射亢进。双足内翻,跟腱挛缩,双侧踝阵挛阳性。脑膜刺激征阴性,共济运动查体不合作。
辅助检查:
血常规、生化全套、甲状腺功能全套、风湿三项、贫血筛查、铜蓝蛋白均未见明显异常。
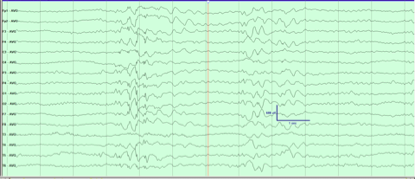
视频脑电图:监测中各区可见短阵暴发中-高幅持续约 2-10 秒的θ和δ活动及棘尖慢波。
颅脑 MRI:T2WI 示双侧尾状核、豆状核对称性萎缩,DWI 未见弥撒受限。
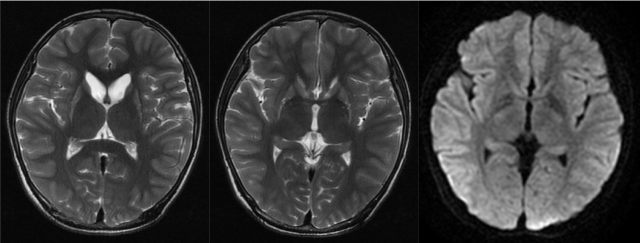
患者主要表现为运动障碍、智能与发育落后、癫痫,定位于基底节和大脑皮层。结合 MRI 表现,定位于双侧纹状体。
双侧纹状体对称性萎缩,需要考虑哪些疾病? 下一步检查?
1. 双侧基底节对称性受累的疾病分类
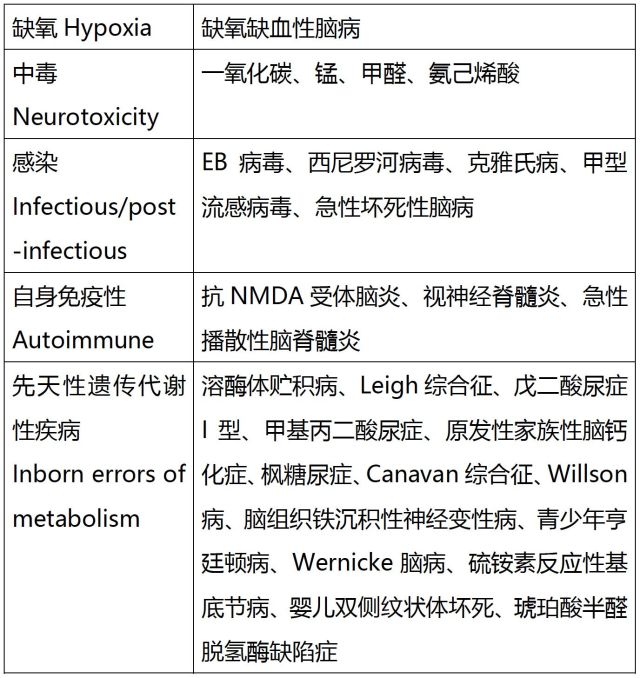
该患者无缺血缺氧病史,无药物及毒物接触史。详细追问患者家属,告知其奶奶成年后出现舞蹈样动作(可疑家族史?)。
结合上表,病因考虑为先天性遗传代谢性疾病可能。
2. 双侧纹状体病变临床诊断思路
双侧纹状体病变可根据影像学表现是否为纹状体坏死缩小诊断范围。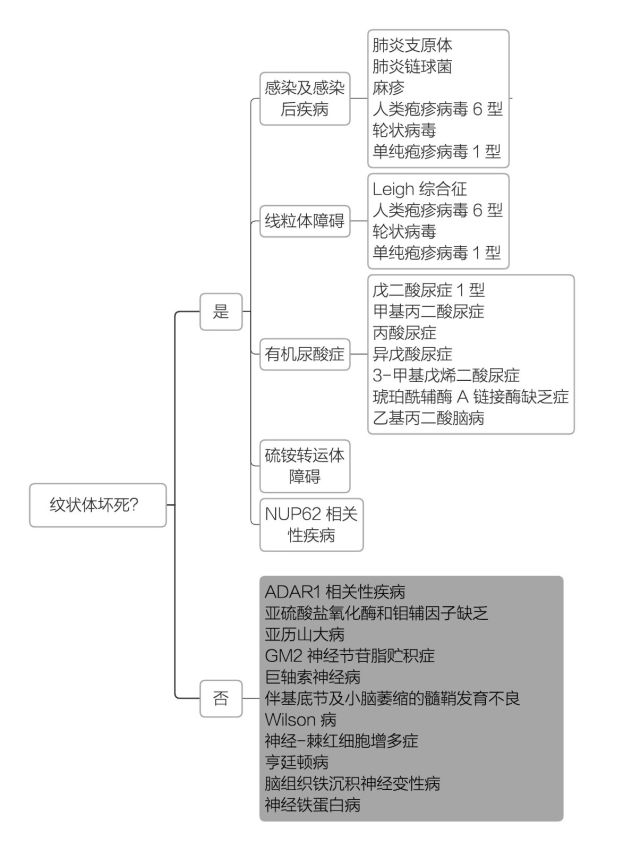
患者影像学提示双侧纹状体对称性萎缩而无坏死(空洞形成)表现,最终将病因锁定至上图灰色区所列出的先天性遗传代谢性疾病。
我院完善外周血涂片未见棘红细胞、裂隙灯下未见 K-F 环及铜蓝蛋白正常,排除神经-棘红细胞增多症和 Wilson 病。
因家庭经济受限,目前临床和影像考虑青少年亨廷顿病可能性大,暂时只选择外送亨廷顿病基因检测。
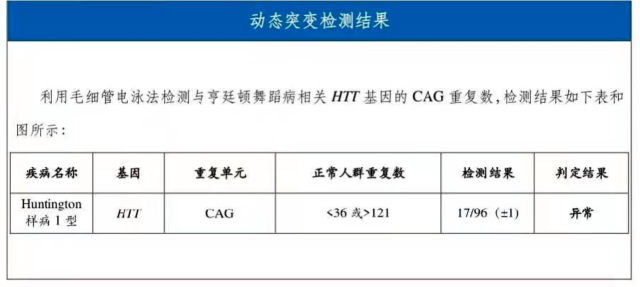
20 天后回报检查结果最终确诊为青少年型亨廷顿病。
青少年型亨廷顿病
亨廷顿病(Huntington disease, HD)是一种以神经系统退行性病变为特征的常染色体显性遗传性疾病。临床以舞蹈样动作、认知障碍和精神障碍为主要特征。
HD 多成人起病,平均发病年龄为 40 岁。然而,有 4%~10% 的患者在青少年期(通常在 20 岁之前)起病, 称之为青少年型亨廷顿病。
青少年型与成人型之间的差异:
回顾一下 2018 年发表在 Lancet Neurol 的一篇关于青少年型亨廷顿病的评论。儿童和青少年起病的 HD CAG 重复拷贝数在 41-174 之间,起病年龄在 2-20 岁之间;而成人型 HD CAG 平均重复拷贝数为 43,往往诊断在 40-50 岁之间。
Fusilli 等将 36 例青少年 HD 患者分为两组:①10 例儿童纳入高扩增突变组(HE 组,CAG 重复数:80-174);②26 例儿童纳入低扩增突变组(LE 组,CAG 重复数:41-73)。
在青少年期间两组以帕金森综合征和肌张力障碍常见。HE 组步态异常多见,而 LE 组以手部灵活度下降多见。随着时间推移两组差别更大。HE 组以步态异常、发育落后、癫痫常见,而 LE 组以舞蹈样动作和行为异常多见。HE 组中位生存期较 LE 组短,提示其进展更快 [3, 4]。
双侧纹状体异常的疾病
1.Aicardi-Goutières 综合征(AGS)
AGS 是一组罕见的以神经系统及皮肤受累为主的遗传性疾病,主要临床特征包括颅内多发钙化灶、脑白质病变、脑脊液慢性淋巴细胞增多症和冻疮样皮损。
至今已发现 7 个 AGS 致病基因,包括 TREX1、RNASEH2B、RNASEH2C、RNASEH2A、SAMHD1、ADAR1 和 IFIH1。
ADAR1 突变阳性患者 MRI 可显示双侧尾状核和壳核对称性信号改变,晚期出现萎缩。
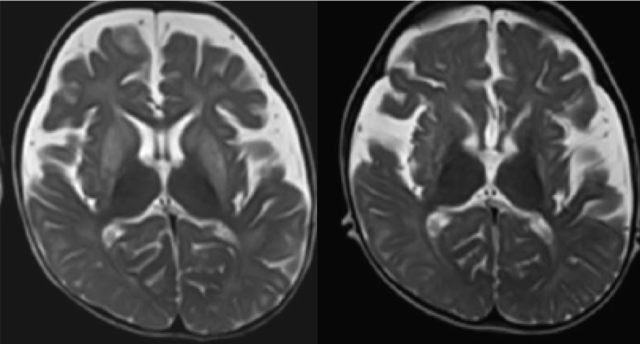
图源:参考文献 2
2. 亚历山大病 I 型
该病是由编码星形胶质细胞特有的胶质纤维酸性蛋白基因突变导致的罕见常染色体显性遗传性白质脑病。临床表现为精神运动发育迟滞、头围增大、癫痫发作、脑干症状、共济失调及神经系统的退行性病变等。
I 型幕上白质、基底节、脑干易受累。亦有影像学脑白质营养不良缺如的报道。其萎缩过程缓慢且于全脑萎缩平行。
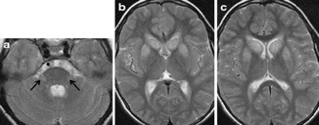
图源:参考文献 2
3. Wilson 病
是一种少见的常染色体隐性遗传性铜代谢障碍性疾病。多青少年起病,临床表现为肝病、神经系统症状和精神症状。
早期磁共振可以无异常表现或者显示新纹状体萎缩,可伴有苍白球、丘脑、脑干及白质损害。很多患者仅表现为新纹状体受累。
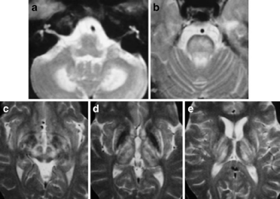
图源:参考文献 2
4. 舞蹈病-棘红细胞增多症:
是一种十分罕见的累及多系统的常染色体隐性遗传性疾病(染色体 9q21 的 VPS13A 基因突变)。表现为基底核退行性病变伴有外周血涂片棘红细胞增多。临床症状主要有运动障碍,认知功能减退,精神异常等。
影像学显示双侧纹状体萎缩。
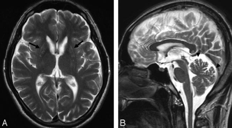
图源:参考文献 5
5. 硫铵素反应性基底节病:
是一种少见的常染色体隐性遗传性疾病,由编码硫铵转运体的 SLE19A3 基因突变所致。该病好发于儿童,表现为脑病、癫痫和锥体外系症状。
磁共振表现为尾状核、壳核、丘脑中间核、皮层核背侧脑干 T2WI 高信号。慢性期则表现为受累区域萎缩和胶质增生。
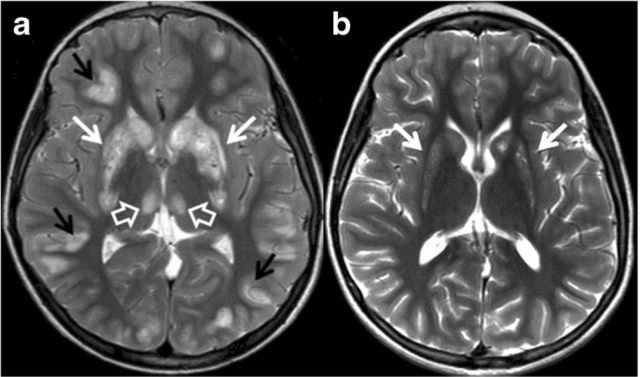
图源:参考文献 1
作者:九江学院附属医院神经内科 沈遥遥 柴竞艳 何云霞 聂红兵
参考文献
[1] Zuccoli G, Yannes MP, Nardone R, Bailey A, Goldstein A. Bilateral symmetrical basal ganglia and thalamic lesions in children: an update (2015). Neuroradiology 2015, 57(10).
[2] Tonduti D, Chiapparini L, Moroni I, Ardissone A, Zorzi G, Zibordi F, et al. Neurological Disorders Associated with Striatal Lesions: Classification and Diagnostic Approach. Curr Neurol Neurosci Rep 2016, 16(6).
[3] Stout JC. Juvenile Huntington’s disease: left behind? Lancet Neurol 2018 11;17(11).
[4] Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, et al. Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol 2018 11;17(11).
[5] Katsube T, Shimono T, Ashikaga R, Hosono M, Kitagaki H, Murakami T. Demonstration of Cerebellar Atrophy in Neuroacanthocytosis of 2 Siblings. AJNR 2009, 30(2).
